碳氧𬭩离子
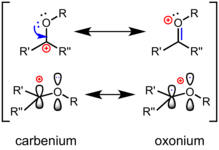
碳氧𬭩离子(或氧碳𬭩离子)是一类在中心sp2-杂化的碳原子上连有氧取代基,并且能够在中心碳原子和氧原子之间通过Π键离域分散所携带的正电荷的化学物种。 1 碳氧𬭩离子可以用两个共振极限结构表示,一个是碳正离子形式,另一个是满足8电子规则且氧带形式电荷的氧𬭩形式。 真实结构为共振杂化体,介于两者之间。碳氧𬭩离子与酮或酯等中性羰基化合物相比,碳正离子形式的共振式对共振杂化体的结构的贡献更大。碳氧𬭩离子是糖苷键水解中常见的反应性中间体,并且是糖基化合成的常用策略。 这些离子作为多种化学反应中的活性中间体,已在有机合成中得到广泛应用,且已成功用于几种天然产物的全合成中。 此外,它们通常在自然界中以酶催化下的生物化学合成和碳水化合物水解的机理出现。 花青素是天然的黄酮染料,是稳定的氧碳𬭩化合物。 花色苷是造成多种常见花(例如紫罗兰)和食用植物(例如茄子和蓝莓)显现颜色的原因。
电子分布和反应性
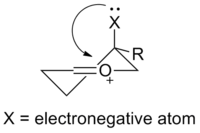
氧碳𬭩离子的最优共振式包含一个碳氧双键 ,该氧原子与另一个基团相连,因此带有形式正电荷。 经典价键理论(或“共振式”)认为,π键的极化可以用次稳定的共振式描述,碳周围有六个电子且携带形式正电荷(见上文)。 根据前线分子轨道理论 ,碳氧𬭩离子的最低未占据分子轨道 (LUMO)是π *轨道,在碳原子上具有较大波瓣。电负性较高的氧对LUMO的贡献相对较小。 因此,在发生亲核进攻时 ,碳是亲电位点 。 与酮相比,碳氧𬭩离子双键的极化作用更加突出:它们更像“真正的”碳正离子,并且对亲核试剂更具反应性。 在有机反应中,通常通过路易斯酸或布朗斯台德酸与氧的配位来活化酮,生成氧碳𬭩离子中间体。
在数值上,酮R2C=O(如丙酮)的羰基碳所携带的的典型部分电荷为δ+ = 0.51(源自Hartree-Fock计算)。而当酮的氧原子质子化后产生[R2C=OH]+ 物种,部分电荷增加至δ+ = 0.61。 相比之下,酮和碳氧𬭩离子的氮取代类似物,亚胺(R2C=NR)和亚胺𬭩离子([R2C=NRH]+)的部分电荷分别为δ+ = 0.33和δ+ = 0.54 。因此,羰基碳所携带的部分正电荷大小的顺序为亚胺<酮<亚胺𬭩<碳氧𬭩。
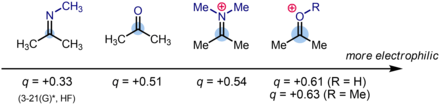
这也是包含 C=X (X = O, NR)键的物种的亲电性的顺序。 这个顺序在有机合成中得到了应用和解释,例如:为什么还原胺化通常最好在pH= 5至6时进行,这个反应用到了 氰基硼氢化钠 (Na+[H3B(CN)]−) 或三乙酰氧基硼氢化钠(Na+[HB(OAc)3]−)作还原剂。 带有吸电子基团的氰基硼氢化钠和三乙酰氧基硼氢化钠比硼氢化钠还原能力弱,它们与酮的直接反应通常是一个缓慢而低效的过程。 但是,在还原胺化反应过程中原位形成的亚胺𬭩离子(并非是亚胺本身)比酮类原料具有更强的亲电性,并且将以在合成中有价值的速率与负氢还原剂反应。 重要的是,该反应是一般在弱酸性条件下进行的,此时电中性的亚胺中间体显著质子化,形成亚胺𬭩离子,而在弱酸性环境中,酮并不能够质子化,亲电性依然较弱。 因此,反应条件和试剂确保了在该反应中由亚胺基还原选择性地形成胺,而不是直接还原羰基(或其质子化形式)以形成醇。
形成
氧碳𬭩离子的形成可以通过几种不同的途径进行。 最常见的是:酮的氧原子与路易斯酸配位,从而活化酮,使其成为更有效的亲电子试剂。 路易斯酸可以是多种分子或离子,可以是质子或金属配合物等。 本条目的其余部分将重点讨论烷基氧碳𬭩离子。烷基氧碳𬭩离子额外连接在羰基氧上的原子是碳。 形成这种离子的一种方式是离去基团的消除,在糖化学中,该离去基团通常是醚或酯 。
合成应用
五元环
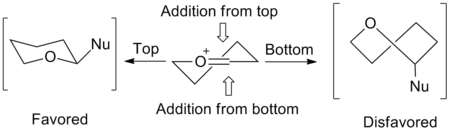
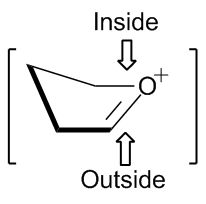 五元环反应所涉及的立体化学可以通过信封形过渡态模型来预测。 亲核试剂有利于从信封的“内部”或右侧图的顶部加成。从信封内部加成将产生有利的交错式构象,而从信封外加成则将产生能量不利的重叠式构象 。 2
五元环反应所涉及的立体化学可以通过信封形过渡态模型来预测。 亲核试剂有利于从信封的“内部”或右侧图的顶部加成。从信封内部加成将产生有利的交错式构象,而从信封外加成则将产生能量不利的重叠式构象 。 2
六元环
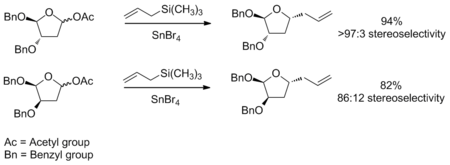
 Woods等人于1992年提出了六元碳氧𬭩环的过渡态模型。 3 确定六元环的亲核加成的立体化学的一般策略与五元环类似。 此分析依据的一种假设是:该环与环己烯具有相同的构象,其中三个碳和一个氧在一个平面内,而另外两个碳原子一个在该平面上方,另一个在下方(请参阅右图)。 依据环上存在的取代基,可以确定最低的能量构象,确定时要考虑空间和立体电子效应。 确定了这种构象后,就可以考虑亲核加成的立体化学。 加成将通过低能量的椅式过渡状态进行。可以在下面看到此类反应的示例。 该示例还说明了负电取代基可能会有较明显的的立体电子效应,通常这会改变最优构象的结构,使得反应的选择性发生变化。 4
Woods等人于1992年提出了六元碳氧𬭩环的过渡态模型。 3 确定六元环的亲核加成的立体化学的一般策略与五元环类似。 此分析依据的一种假设是:该环与环己烯具有相同的构象,其中三个碳和一个氧在一个平面内,而另外两个碳原子一个在该平面上方,另一个在下方(请参阅右图)。 依据环上存在的取代基,可以确定最低的能量构象,确定时要考虑空间和立体电子效应。 确定了这种构象后,就可以考虑亲核加成的立体化学。 加成将通过低能量的椅式过渡状态进行。可以在下面看到此类反应的示例。 该示例还说明了负电取代基可能会有较明显的的立体电子效应,通常这会改变最优构象的结构,使得反应的选择性发生变化。 4
立体电子效应
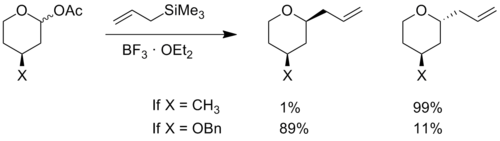
在不包含氧原子的烯烃环中,任何大的取代基都倾向于位于赤道位置,以使空间效应最小化。 但是在含碳氧𬭩离子的环中, 负电性取代基倾向于优先占据轴向或准轴向位置。 当负电性原子处于轴向位置时,其电子密度可以通过立体电子效应分散给环中带正电的氧原子。 5 这种相互作用稳定了轴向构象。表现出这种现象的取代基的实例主要有 羟基 , 醚和卤素。 在确定氧代碳𬭩离子的亲核加成反应中能量最低的构象时,必须考虑立体电子效应 。 4 6
环加成
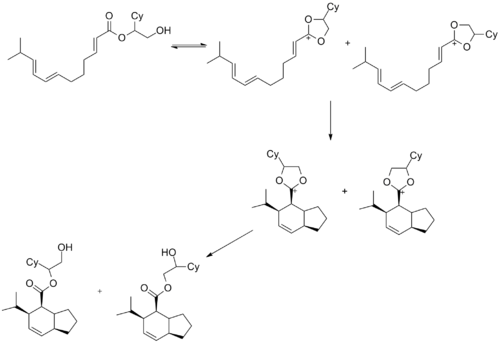
在有机合成中,乙烯基氧碳𬭩离子(右侧结构)可广泛用于环加成反应中。 它们通常在Diels-Alder反应中用作亲二烯体 。 通常将吸电子酮添加到该亲二烯体中以提高反应速率, 7在反应过程中,这些酮经常转化为乙烯基氧碳𬭩离子8。目前尚不清楚该机理是否必须经历碳氧𬭩离子中间体,但Roush和他的同事在以下所示的环化反应中证明了该反应确实生成了氧碳𬭩离子中间体。 该反应可观察到的两种产物只有在氧碳𬭩环作为中间体存在时才能形成。已经有人报道氧碳𬭩离子中间体参与的9 [4+3], [2+2], [3+2]和[5+2]环加成。
羟醛反应
已有利用手性氧碳𬭩离子与乙烯醇衍生等价物进行高度非对映选择性和对映选择性的反应 10 氧碳𬭩离子在反应中用作亲电试剂 。 当甲基替换为更大的取代基时,反应的非对映选择性增加。
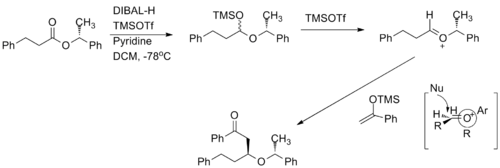
全合成实例
氧碳𬭩离子已在多种场合中用于全合成。(+)-clavosolide的主要亚基是通过还原六元氧碳𬭩环合成的。 如所预测的,所有大的取代基都位于赤道位置,并且该转化经历了椅式过渡状态。 11
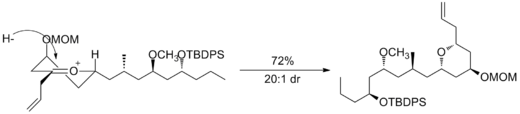
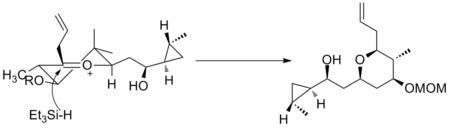 第二个例子可见于 (−)-neopeltolide合成的关键步骤,该步骤使用另一种六元氧碳𬭩环发生还原反应生成非对映选择性加氢产物。 12
第二个例子可见于 (−)-neopeltolide合成的关键步骤,该步骤使用另一种六元氧碳𬭩环发生还原反应生成非对映选择性加氢产物。 12
生物学应用
在生物系统中,氧碳𬭩离子最常见于碳水化合物的反应过程中。 由于糖存在于核酸中,它的化学性质对核酸在生物体内的功能起到了重要作用。 糖除了在核酸中发挥生理功能外,还是生物体的结构组分,可以多种形式存在,例如储能分子、细胞信号分子、蛋白质修饰,并在免疫系统 、 受精作用 、预防发病机制 、血液凝结和发育中发挥关键作用。 13 生物化学过程中糖化学性质丰富,许多糖参与的反应经历氧碳𬭩离子中间体。 本节概述了利用氧碳𬭩离子的几种重要的生物反应。
核苷酸生物合成
酶催化下核苷酸的分子内环化可产生几种重要的生物分子。 这些环化通常通过氧碳𬭩中间体进行。 该反应的一个例子是环ADP核糖的环化。 14
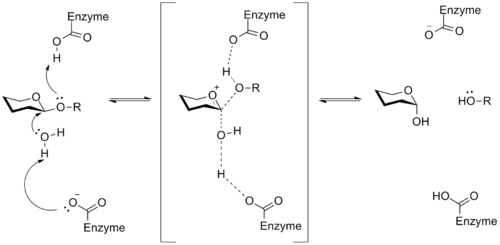
糖苷酶
糖苷酶是一种催化糖苷键断裂的酶,它能使底物糖分解产生两个较小的糖分子 。 这个过程对动物体内糖原等储能物质的利用以及植食生物进行纤维素的分解具有重要意义。 通常,酶的活性位点上的天冬氨酸或谷氨酸残基可以催化糖苷键的水解。 这些酶的机制涉及氧碳𬭩离子中间体,其一般实例如下所示。 15
参见
碳正离子
糖苷酶
本文采用CC-BY-SA-4.0协议发布,来自我创建的Wikipedia条目碳氧𬭩离子,由于个人水平限制,本条目难免会有知识谬误以及描述不清等情况,欢迎读者在维基条目中编辑修改或对本网站在GitHub上的源代码提供Pull request,也欢迎在评论区多多指教。
参考文献
-
(英文)国际纯粹与应用化学联合会.”carbenium ion”.《化学术语总目录》在线版. ↩
-
Larsen, C. H.; Ridgway, B. H.; Shaw, J. T.; Woerpel, K. A., A Stereoelectronic Model to Explain the Highly Stereoselective Reactions of Nucleophiles with Five-Membered-Ring Oxocarbenium Ions, Journal of the American Chemical Society, 1999, 121 (51): 12208–12209, doi:10.1021/ja993349z ↩
-
Woods, R. J.; Andrews, C. W.; Bowen, J. P., Molecular mechanical investigations of the properties of oxocarbenium ions. 2. Application to glycoside hydrolysis, Journal of the American Chemical Society, 1992, 114 (3): 859–864, doi:10.1021/ja00029a008 ↩
-
Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A., Stereochemical Reversal of Nucleophilic Substitution Reactions Depending upon Substituent: Reactions of Heteroatom-Substituted Six-Membered-Ring Oxocarbenium Ions through Pseudoaxial Conformers, Journal of the American Chemical Society, 1999, 122: 168–169, doi:10.1021/ja993366o ↩ ↩2
-
Miljkovic, M. i.; Yeagley, D.; Deslongchamps, P.; Dory, Y. L., Experimental and Theoretical Evidence of Through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis, The Journal of Organic Chemistry, 1997, 62 (22): 7597–7604, doi:10.1021/jo970677d ↩
-
Hansen, Thomas; Lebedel, Ludivine; Remmerswaal, Wouter A.; van der Vorm, Stefan; Wander, Dennis P. A.; Somers, Mark; Overkleeft, Herman S.; Filippov, Dmitri V.; Désiré, Jérôme. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Central Science. 2019-04-18, 5 (5): 781–788. ISSN 2374-7943. doi:10.1021/acscentsci.9b00042. ↩
-
Vollhardt; Shore. Organic Chemistry. New York, NY: W. H. Freeman and Co. 2009. ↩
-
Harmata, M.; Rashatasakhon, P., Cycloaddition reactions of vinyl oxocarbenium ions, Tetrahedron, 2003, 59 (14): 2371–2395, doi:10.1016/s0040-4020(03)00253-9 ↩
-
Roush, M.; Gillis, P.; Essenfeld, A., Hydrofluoric acid catalyzed intramolecular Diels–Alder reactions, Journal of Organic Chemistry, 1984, 49 (24): 4674–4682, doi:10.1021/jo00198a018 ↩
-
Kanwar, S.; Trehan, S., Acetate aldol reactions of chiral oxocarbenium ions, Tetrahedron Letters, 2005, 46 (8): 1329–1332, doi:10.1016/j.tetlet.2004.12.116 ↩
-
Carrick, J. D.; Jennings, M. P., An Efficient Formal Synthesis of (−)-Clavosolide a Featuring a “Mismatched” Stereoselective Oxocarbenium Reduction, Organic Letters, 2009, 11 (3): 769–772, doi:10.1021/ol8028302 ↩
-
Martinez-Solorio, D.; Jennings, M. P., Formal Synthesis of (−)-Neopeltolide Featuring a Highly Stereoselective Oxocarbenium Formation/Reduction Sequence, The Journal of Organic Chemistry, 2010, 75 (12): 4095–4104, doi:10.1021/jo100443h ↩
-
Maton, Anthea; Jean Hopkins; Charles William McLaughlin; Susan Johnson; Maryanna Quon Warner; David LaHart; Jill D. Wright. Human Biology and Health. Englewood Cliffs, New Jersey, USA: Prentice Hall. 1993: 52–59. ISBN 0-13-981176-1. ↩
-
Muller-Steffner, H. M.; Augustin, A.; Schuber, F., Mechanism of Cyclization of Pyridine Nucleotides by Bovine Spleen NAD+ Glycohydrolase, Journal of Biological Chemistry, 1996, 271 (39): 23967–23972, PMID 8798630, doi:10.1074/jbc.271.39.23967 ↩
-
Zechel, D.L.; Withers, S.G. Glycosidase Mechanisms: Anatomy of a Finely Tuned Catalyst. Accounts of Chemical Research. 2000, 33 (1): 11–18. ISSN 0001-4842. PMID 10639071. doi:10.1021/ar970172. ↩
赞赏本文
| 支付宝 | 微信支付 |
|---|---|
 |
 |